4月26日,加拿大健康科学院院士,温州医科大学学术副校长、老年研究院院长,瓯江实验室主任宋伟宏院士团队在Nature Communications《自然·通讯》发表了题为“Diphthamide deficiency promotes association of eEF2 with p53 to induce p21 expression and neural crest defects”的研究论文,发现白喉酰胺生物合成1(Diphthamide biosynthesis 1, DPH1)基因突变在儿童罕见病“发育迟缓、身材矮小和头发稀疏综合征”(Developmental delay, short stature, and sparse hair syndrome, DEDSSH)中的致病机制,为DEDSSH及认知和发育障碍相关疾病的药物开发和治疗提供了新靶点。

DEDSSH是一种罕见的儿童发育障碍综合征,其特征包括不同程度的认知功能障碍、发育迟缓、身材矮小、形态特征异常以及头发稀疏等症状,目前全球仅有几十例病案报道。DEDSSH是一种常染色体隐性遗传疾病,通常由DPH1基因突变所引起,故也称之为DPH1综合征。DPH1基因位于人类染色体17p13.3上,由13个外显子组成,其编码生成由438个氨基酸组成的合成白喉酰胺的关键酶。白喉酰胺是一种独特的经过翻译修饰后的组氨酸,仅存在于古菌和真核生物的延伸因子eEF2中。DPH1是催化eEF2中位于715的组氨酸修饰为白喉酰胺所必需的酶。白喉酰胺对eEF2的翻译后修饰具有高度保守性,该修饰过程对eEF2在蛋白质合成过程中的正常功能维持至关重要。eEF2与神经发育和突触可塑性及认知功能密切相关。但DPH1基因突变在DEDSSH患者中白喉酰胺异常修饰引发认知和发育障碍的具体机制不清。
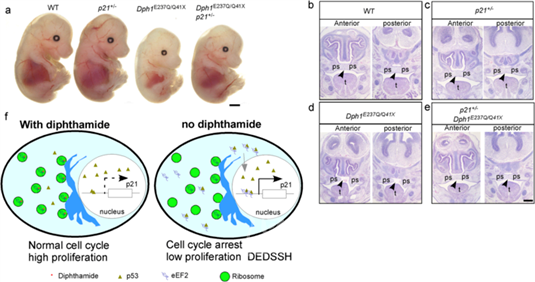
在本项目前期,宋院士团队发现一个4岁的女性儿童DEDSSH患者,她分别从父亲和母亲遗传到DPH1基因的两个不同的杂合突变,表现出全身性发育迟缓和认知障碍等临床表型,其父母由于仅携带1个DPH1基因的杂合突变,所以并未表现出该病的临床症状。本研究通过对DEDSSH患者的淋巴系细胞的检测,发现白喉酰胺修饰的eEF2水平降低与患者临床表型密切相关。实验证明DPH1基因缺失抑制了爪蟾模型中神经嵴前体细胞的增殖。团队构建了携带患者DPH1突变基因敲入的模型小鼠(Dph1E237Q/Q41X),发现Dph1-E237Q和Dph1-Q41X共突变显著降低了白喉酰胺修饰的eEF2水平,进而导致Dph1E237Q/Q41X突变小鼠呈现出DEDSSH认知障碍和颅面部畸形等临床表型,表明DPH1基因突变引发的白喉酰胺修饰异常对DEDSSH发病起着关键作用。此研究进一步发现白喉酰胺修饰的eEF2水平降低可诱导eEF2-p53复合物形成增多,促进了p53与p21基因启动子区域的结合,进而诱导p21的转录水平升高,导致神经嵴在内的神经内皮细胞的增殖发生异常;而降低P21的表达水平可明显改善Dph1E237Q/Q41X突变基因敲入小鼠的表型。这项研究阐明了白喉酰胺修饰调控的eEF2作为p53的转录共激活因子引发神经发育缺陷中的重要作用,揭示了DPH1基因突变导致儿童罕见发育障碍综合征的致病新机制,为认知和发育障碍相关疾病的药物开发和治疗提供了新靶点。
我校宋伟宏院士、重庆医科大学附属儿童医院石宇副研究员和University of Delaware Shuo Wei教授为本项研究成果的共同通讯作者。宋伟宏院士团队的石宇副研究员、黄道超博士和宋萃主任医师为文章的共同第一作者。
扫一扫在手机打开当前页




